
科学家发展“辣搜”预测化学无序材料的电子结构
- 2023/2/20 9:17:20 19348
- 来源:中国科学院力学研究所
【仪表网 研发快讯】 确定材料的微结构是认识和改性材料的前提。化学无序材料指晶格有序但元素成分无序的一类材料。从化学成分的角度来看,化学无序材料可分为阴离子、阳离子和缺陷对应体,可以简单地认为是阴离子、阳离子和缺陷占据了非周期位点。化学无序材料由于其独特的性质在半导体、高温超导体、金属合金、陶瓷和沸石催化剂等领域被广泛应用。
研究无序材料的结构对于理解无序材料性质和指导实验具有重要价值。因为部分晶格位点的原子占据不确定性,化学无序材料的确切结构一直尚未解决。化学无序材料的结构预测是极具挑战的,其随着体系尺寸增加变得愈加复杂。前期研究利用晶体对称性除重、集团展开和机器学习势等技术加速了化学无序材料的结构预测,但仍面临大量昂贵的第一性原理计算。
中国科学院力学研究所和中科院山西煤炭化学研究所/中科合成油技术有限公司联合团队发展了“辣搜”(LAsou)方法来高效预测化学无序材料的电子结构。三种不同的、典型的有限尺寸体系测试表明【阴离子无序的BaSc(OxF1-x)3、阳离子无序的Ca1-xMnxCO3和缺陷无序的ε-FeCx】,与传统枚举法相比,“辣搜”方法仅需要非常少的第一性原理计算就可以快速找到热力学稳定的结构。
“辣搜”方法是一种简单而高效的方法,结合第一性原理计算和主动学习算法来搜索化学无序材料的热力学稳定结构。“辣搜”方法在解决多体体系的“指数墙”(由于计算复杂度随自由度指数上升而形成的壁垒)问题方面显示出巨大潜力。
在“辣搜”方法中,机器学习势可以通过大采样空间的预测和筛选大大减少第一性原理的计算量,集成学习算法可以显著提高能量和弛豫预测的稳定性,主动学习算法可以在线逐步提高机器学习势的精度,从而不需要预先准备大量的训练数据。结合这些优点和特点,基于主动学习的“辣搜”方法将有助于更大、更复杂、准无限尺寸系统的广泛应用,以及出现在纳米颗粒、催化剂、固溶体、高熵合金和高熵氧化物等新材料中。

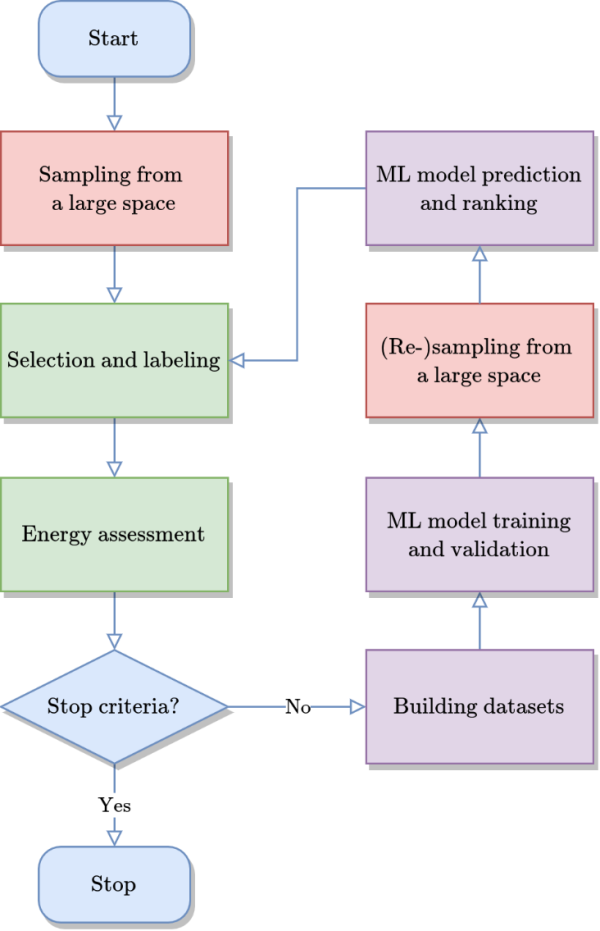
图1 基于主动学习的“辣搜”方法流程图。概述了与第一性原理计算相结合的用于有限尺寸化学无序材料结构预测的方法。

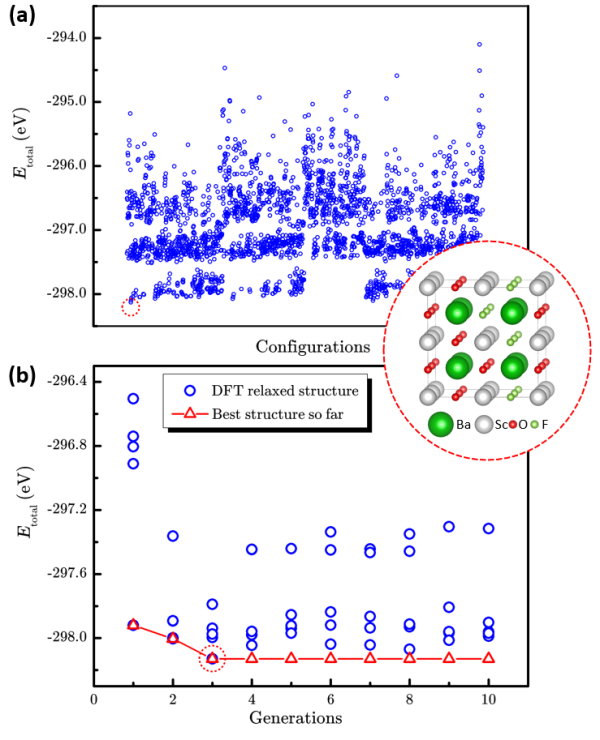
图2 枚举法和“辣搜”法在BaSc(OxF1-x)3 (x=0.667)体系的表现。(a)2664个枚举结构和最稳定结构的总能量散点图。(蓝色圆圈代表每个结构的能量,红色虚线圆圈代表能量最低的结构。)(b)“辣搜”方法搜索过程中总能量随搜索代数的演化。(红色三角形代表历史上搜索过的最低能量结构。)

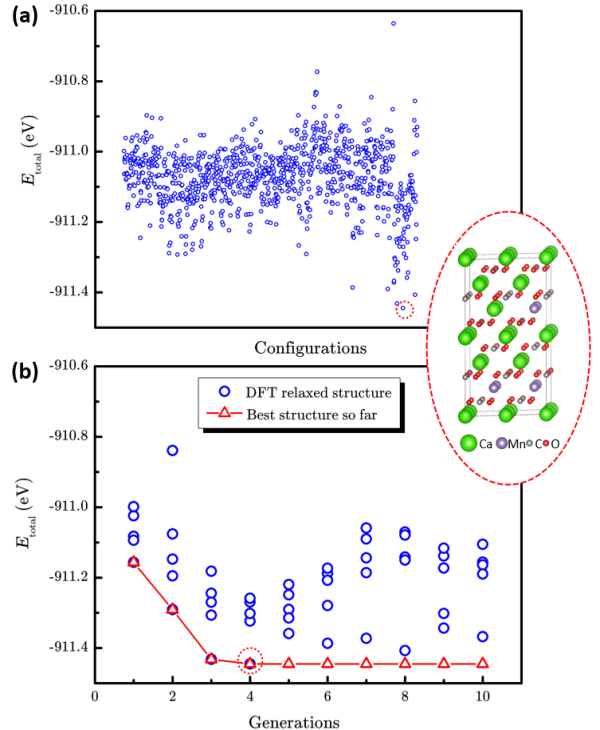
图3 枚举法和“辣搜”法在Ca1-xMnxCO3 (x=0.25)体系的表现。(a)1033个枚举结构和最稳定结构的总能量散点图。(b)“辣搜”方法搜索过程中总能量随搜索代数的演化。

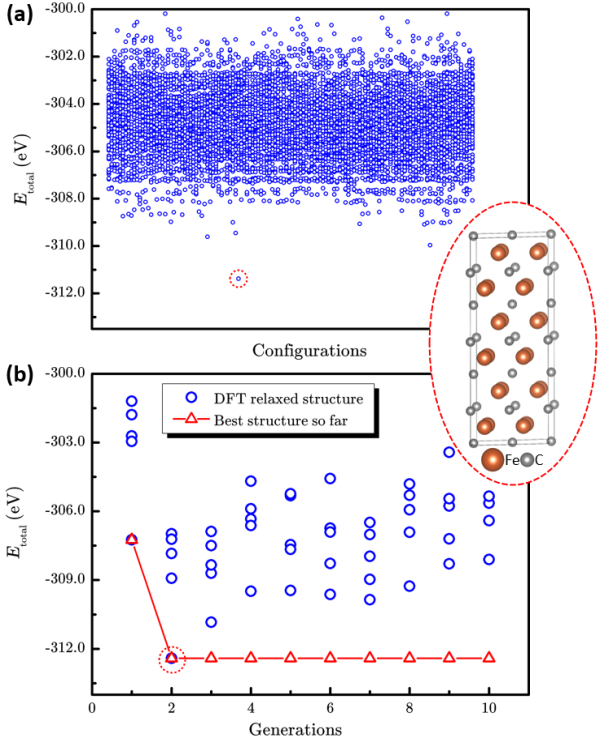
图4 枚举法和“辣搜”法在ε-FeCx (x=0.5)体系的表现。(a)10496个枚举结构和最稳定结构的总能量散点图。(b)“辣搜”方法搜索过程中总能量随搜索代数的演化。



















全部评论